图5 企业生命发展周期
组织对内部运营管理要求为:形成能够有效支持到整合“新”资源、在“新”的环境中,“快速”、“高效”地达成目标的运作模式。
3. 寻找组织的价值点
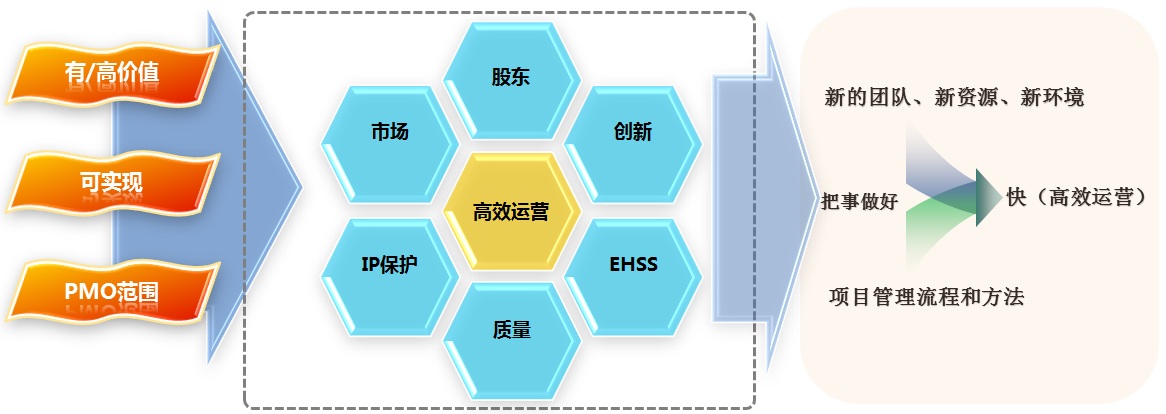
图6 组织落地的价值点
寻找对组织有价值的运营环节、区域,首先,要定义落地的价值点的衡量标准。PMO从三个方面进行考虑。首先是对组织无价值、低价值、有价值且为高价值的运营区域或者环节进行区分,如果这些落地点落在无价值或者低价值区,将不会对组织的运营做出显性、关键贡献。其次,这些价值点是能够实现的。如果从资源或者PMO实现的角度不能实现或者实现难度很高,再有价值的落地点也会没有意义。第三,这些价值的落地点必须是PMO当前的工作范围,也就是需要和PMO管辖的项目(集)相关。如果这些落地点虽然有高价值,但是不是项目(集)所涉及管理的范围,属于其他业务部门的业务范围,价值的依赖性过多或者根本无关,就很难体现PMO的相关作用。
如图6所示,在覆盖组织股东、市场、IP保护、质量、EHSS(环境、健康、安全、安保)、创新等主要运营环节中,选择了“高效运营”这个落地点。首先,该点与组织的主要运营环节都相关。其次,在组织完成架构建设后,“高效运营”贯穿于组织整个生命周期中,属于持续改进的重要目标和组成部分。因此选择“高效运营”作为落地点具有价值高、生命周期长且可实现的优势。
4. 规划行动、实现价值
在开始规划行动之前,需要对关键项进行分析。这些关键项对PMO在组织中生存和发展至关重要。关键项包括:
1) 了解组织内部门合作的本质。
2) 优化跨部门合作及与供应商合作的方式。
3) 形成适合组织、组织适用的项目管理方法(论),结合PMI和V-Model在项目中平衡合规和效率的问题。
同时在GMP环境下,PMO需要解决三个层面的问题:
1) 原非GMP人员不理解项目中GMP要求。
2) GMP环境人员不理解项目管理的要求。
3) GMP环境中结合原有管理系统的利用和优化 (如CC, Deviation,CAPA等)。
如何真正地规划行动协助组织实现价值,如图7所示。
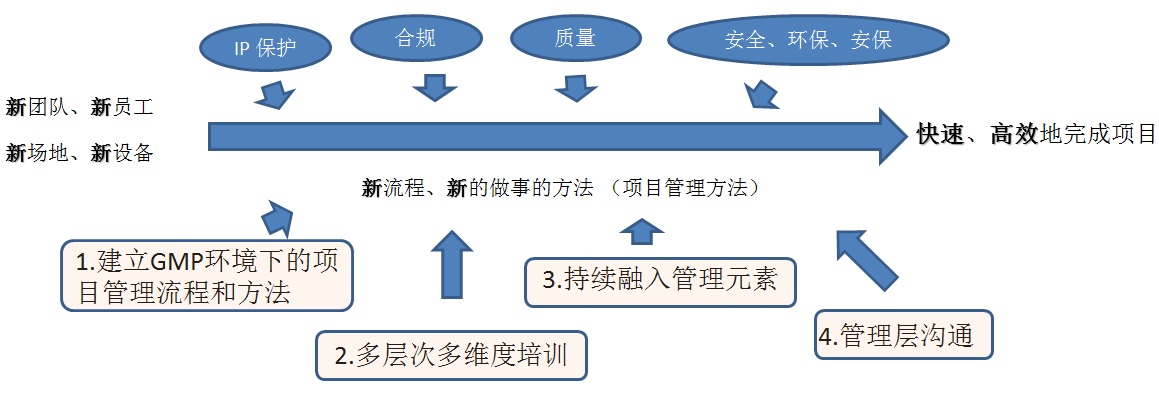
图7 实现价值四个步骤
在组织快速扩展的阶段,组织需要面对的是新团队、新员工、新场地(工厂)和新设备等一个全新的环境。对于一个新团队,在IP保护、合规、质量、安全、健康、环保、安保等方面的要求本身就是一个挑战,同时要达到快速、高效地完成项目的要求,需要有合适的流程、方法协助最终达成目标。具体行动实践参照后续“PMO落地实践”章节。
2.2 PMO的落地实践
2.2.1 组织内部的GMP环境介绍
1. GMP环境概述
了解组织内部的GMP的生产运营环境是PMO在组织能够落地的最基础和重要一步。所在组织内部是一个由合规、审计、风险、质量驱动的运营环境。
合规驱动指的是参照生物制药行业的法律、法规及相关的指南。相关参考文件包括:CFDA《计算机化系统》附录、EUGMP附录11、联邦法规21章11款电子记录、电子签名框架结构、ISPE(GAMP5)、EU药事法规第4章《文件管理》等。
审计驱动指的是由各类官方审计、客户审计等驱动、指导的生产运营过程,如FDA(美国食品药品管理局)、CFDA(中国食品药品管理局)、EMA(欧洲药品管理局)、WHO(世卫组织)、客户审计(如:GSK、Roche)等。
IP保护是组织业务经营秉承的重要原则之一,CMO是为制药行业的企业提供研发、测试及生产等相关服务,对IP的保护是遵守行业规则/获得客户信任的最基本要求。在服务过程中,会接触到客户的配方、专利等商业敏感信息,对于客户信息的保护及对服务过程中产生的数据信息需要全方位进行保护。从商业的合同的条款、到实验人员的培训,以及保密合同和组织内部IT对信息的严格管控,形成了IP保护的若干道防火墙。
风险管理保障了组织的运营活动,在生物医药企业,风险管理是所有运营活动的基础。由于医药制造行业的特殊性,任何活动都需要评估风险对病患、生产过程的稳定性、产品的质量等影响,只有经过评估才能执行活动,活动结束后还要进行风险再评估和确认。
成本和效率是任何组织运营必须考虑的问题。在生物制药行业,其特殊性在于必须符合GMP的要求,要求即成本,如何在合规性要求下去平衡GMP要求和运营效率,是组织面临的挑战,同样也是机遇,因为所有行业的企业都需要考虑这样的平衡。同时组织对成本、效率和合规平衡的要求,为PMO的生存和发展提供了契机。
质量是组织的生命线,是赖于生存和发展的基础。尤其是在药企,其产品的使用对象是病患,产品质量涉及人的生命安全。因此,在组织内部有严格的质量控制体系,主要包括研发的质量、生产的质量和服务的质量。针对组织内部不同的生产经营环节,从上游到下游设置了不同的质量管理控制体系及方法,以保证各环节及整体都符合质量管理的要求。
2. 常用内部质量管理方法
在以上多约束条件的GMP运营环境下,在组织内部使用严格的质量管理手段对所有生产运营涉及的操作进行管理,常用的质量管理方法包括标准操作流程 (Standard Operation Procedure,SOP),偏差管理(Deviation Management,DV),变更控制管理 (Change Control,CC),纠正措施和预防措施 (Corrective Action Preventive Action,CAPA),风险评估(Risk Assessment), 验证和放行(Validation and Release),周期性回顾(Periodic Review )等。
SOP标准操作流程的制定是对生产运营涉及的操作进行标准化、规范化,同时进行指导,并要求严格遵循和执行,如有与标准偏离的情况即判定为偏差(Deviation)。一旦发现或成为偏差,马上对偏差造成的对生产过程及产品产生的影响进行分类评估,同时对偏差原因进行分析,从人、机、料、法、环等方面找到根本原因、直接原因,并形成相对应的纠正和预防措施(CAPA)。每一个偏差由指定的部门起草,并和其他相关部门合作完成调查、影响分析、原因分析、后续纠正和预防措施的指定和完成,并验证效果,由验证和质量部门确认效果、符合质量管理流程并审批后才能关闭,整个偏差的生命周期才算结束。质量体系管理部门对偏差处理的每个环节上(如调查、原因分析、处理措施执行等)都有严格的时间要求,一旦超过了时间,偏差处理本身就形成了偏差,这类偏差同时需要按偏差处理的流程进行处理。
关于变更控制管理(Chang Control,CC)。变更是事物、环境从一个稳态变化到另一个稳态的过程。在生物制药行业中,稳态是一个经过验证、风险受控的环境,从而能够保证生产过程及产品的稳定性。而在一个稳态环境中加入新的元素,如业务需求添加一台新的设备,在GMP环境中就需要进入一个新的稳态,该环境要重新进行风险评估,根据风险评估的级别,需要再次验证来确保该新环境对生产过程和产品无影响或低风险受控。如项目就是一个变更。
关于数据完整性(Data Integration,DI)。数据完整性是对制药行业的统一、最基本和严格的要求。所有的官方审计如美国的FDA 审计、欧盟的EMA审计、中国SFDA食药监的审计是必审的环节。所有药品生产过程中产生的数据包括系统电子签名等必须完整可追溯,以确保一旦药品出了问题,马上可以查出哪个环节出问题。






